
Рассылка
Будьте в курсе последних обновлений – подпишитесь на рассылку материалов на Ваш e-mail
Подписаться-
Всесвітній день поширення інформації про аутизм: спростовуємо поширені міфи
-
Резистентна до лікування депресія: можливості аугментації терапії
-
Фармакотерапія тривожних розладів і нейропротекція: альтернатива бензодіазепінам
-
Лікування пацієнтів підліткового віку із шизофренією: ефективність і безпека антипсихотичної терапії
-
Лікування депресії в пацієнтів з ішемічною хворобою серця або ризиком її розвитку
-
Профілактична фармакотерапія епізодичного мігренозного головного болю в амбулаторних умовах
-
Стратегії зниження дозування бензодіазепінів: коли ризики переважають користь
-
Раздел:
Фармакотерапія тривожних розладів і нейропротекція: альтернатива бензодіазепінам
Зміст статті:
- Особливості патогенезу тривожних розладів
- Взаємодія етифоксину з рецепторами ГАМК-А
- Вплив етифоксину на симптоми тривожних розладів
- Анксіолітична активність етифоксину
- Нейропротекторна дія етифоксину на центральну та периферичну нервову систему
- Висновки
Тривожні розлади — велика група різнорідних щодо причин і механізмів розвитку станів: від психічних порушень до перевтоми та реакції на стреси. Поширеність тривожних розладів дуже висока, практично кожна людина хоча б раз у житті відчуває підвищену тривожність, що спричинює дискомфорт. Ознаками тривожних розладів є збудження й очікування небезпеки. Дослідники продовжують виявляти додаткові рівні складності патофізіологічних механізмів цих розладів.
Як відомо, тривожні розлади часто є коморбідними з порушеннями адаптації — це реактивні та переважно короткотривалі стани, пов’язані зі складними життєвими ситуаціями. Їх проявами є підвищена тривожність, пригнічений настрій або поведінкові симптоми, які розвиваються після значних змін у житті людини або досвіду психологічного напруження.
Концепція лікування розладів адаптації була переосмислена в Діагностичному і статистичному посібнику з психічних розладів 5-го перегляду (DSM-5) та Міжнародній класифікації хвороб 11-го перегляду (МКХ-11). Раніше акцент робили на суб’єктивному дистресі та емоційних порушеннях за розладів адаптації, зокрема з погляду тривожної або депресивної симптоматики, що призводило до їх ототожнення з тривожними розладами й депресіями. Через критику щодо валідності діагностичних критеріїв DSM-IV і МКХ-10 для розладів адаптації в останніх версіях цих класифікацій відбулося зміщення в бік їх концептуалізації як розладів, пов’язаних із травмою та стресом. Діагностичні критерії DSM-5 для розладів адаптації охоплюють тимчасові дезадаптивні або патологічні реакції на ідентифіковані стресори або зміни в життєвих обставинах із розвитком симптомів упродовж трьох місяців після пережитого стресу (Nuss etal., 2019).
Нині тривожність, пригнічений настрій і порушення поведінки розглядають як потенційно асоційовані кваліфікатори, а не як специфікатори. Клінічні ознаки описують як такі, що не відповідають події. Окрім внутрішньої природи, стресор необхідно розглядати в межах особистого та міжособистісного контексту, за якого він стався, а також культурних норм. У МКХ-11 пішли ще далі, визначаючи основні клінічні симптоми, а саме: стурбованість стресором або його наслідками, як-от надмірне занепокоєння, повторювані та тривожні думки про стресор або постійні роздуми про його наслідки; нездатність адаптуватися до стресора, що спричиняє значне порушення в особистих, сімейних, соціальних, освітніх, професійних або інших важливих сферах функціонування. Якщо функціонування зберігається, то лише завдяки значним додатковим зусиллям, а якщо є порушення, воно має бути набагато більшим, ніж очікувалося, зважаючи на попереднє функціонування індивіда. Своєю чергою, краще визначення розладів адаптації допоможе ідентифікувати пов’язані з ними біомаркери, асоційовані як із ЦНС, так і з функціонуванням організму людини загалом, на основі психобіології травми та стресора, що сприятиме ефективності лікування. Також важливо вивчити роль γ-аміномасляної кислоти (ГАМК) у патогенезі розладів адаптації, нейроендокринних процесів, запалення, імунітету та оксидативного стресу (Poisbeau etal., 2018; Strain, 2018).
Особливості патогенезу тривожних розладів
вгоруТрадиційна концепція патофізіології підвищеної тривожності ґрунтується на структурній і функціональній дисфункції мозку (Nuss, 2015). Вона розглядає тривожність як наслідок зміни скоординованої активності шляхів мозку, що модулюється локальними й віддаленими «синаптичними реле» через нейромедіатори. Як відомо, до цього процесу залучений набір лімбічних структур, зокрема мигдалеподібного тіла, яке тісно пов’язане з префронтальною корою і, ймовірно, відіграє вирішальну роль у регуляції негативних емоцій. Різні нейротрансмітери та модулятори мають важливе функціональне значення в модулюванні поведінки, пов’язаної з тривожністю. До них належать медіатори, пов’язані з гіпоталамо-гіпофізарно-наднирниковою (ГГН) віссю, моноамінергічні та ГАМК-ергічні системи нейротрансмісії, нейропептиди (як-от холецистокінін) і ліпідні нейромодулятори (Hauger etal., 2006; Mehta and Ticku, 1999; Boyer, 2000; Bandelow etal., 2017).
На додаток до такого орієнтованого на мозок підходу до патофізіології тривожності все більше є доказів на користь того, що її можна модулювати впливом на мозок соматичних фізіологічних процесів, як-от запалення, імунітет і оксидативний стрес, а також функціонування мікробіоти кишківника (Michopoulos etal., 2017). Дисфункціональні взаємодії за участю ГГН-осі та кишкової мікробіоти були описані як такі, що роблять внесок у патофізіологію тривожних розладів (Sharon etal., 2016).
Стрес — спільна риса всіх тривожних розладів — пов’язаний із декількома фенотипами прозапальної відповіді, які можуть не реагувати на протизапальну дію глюкокортикоїдів (Cohen etal., 2012). Деякі препарати, які, як відомо, зменшують клінічні симптоми тривожних розладів (антидепресанти, певні бензодіазепіни [БЗД] та небензодіазепінові анксіолітики), послаблюють вплив зазначених аномальних фізіологічних процесів, що вказує на можливий — і раніше недооцінений — аспект механізму дії анксіолітичних засобів (Ghareghani etal., 2017).
Сучасне розуміння патогенезу тривожних розладів та їх лікування може потребувати розширення і залучення впливу механізмів, не пов’язаних із нервовою системою. Також варто переглянути розуміння механізмів дії анксіолітичних препаратів як у межах ЦНС, так і поза ними.
Етифоксин, небензодіазепіновий анксіолітичний препарат, що діє як позитивний алостеричний модулятор ГАМК-ергічної передачі, не є винятком із цього правила. Він може виявляти значну протизапальну активність у ЦНС, вплив на імунну та нейроендокринну систему, зокрема через зв’язування з білком-транслокатором зовнішньої мембрани мітохондрій (TSPO) і синтез нейротрофічного фактора, а також сприяти анксіолітичній активності вказаного засобу (Simon-O’Brien, 2016; Juif etal., 2015; Do Rego etal., 2015; Girard etal., 2008). Такий підхід відкриває нові перспективи для вивчення механізмів дії цього анксіолітика.
Лікування тривожних розладів базується на доказових даних досліджень, а також емпіричних експериментів із застосуванням ліків. Зважаючи на широке розмаїття клінічних ситуацій, які супроводжуються тривожністю, і хронізації цих ознак нереалістично очікувати єдиного оптимального доказового підходу до лікування для таких ситуацій (Nuss etal., 2019). Клініцистам необхідне глибше розуміння різноманітних механізмів дії анксіолітичних засобів, щоб вибрати перспективний анксіолітик для пацієнтів із тривожними розладами.
Взаємодія етифоксину з рецепторами ГАМК-А
вгоруРоль нейротрансмісії ГАМК-А у патофізіології тривоги
Результати досліджень на тваринних моделях і вивчення нейровізуалізаційних даних підтвердили, що тривала дизрегуляція мереж мозку, яка охоплює коркові та специфічні підкіркові ділянки (мигдалину, гіпокамп, таламус, префронтальну та поясну кору), позначається на виразності ознак тривожних розладів (Nuss, 2015). Зниження гальмівної ГАМКергічної передачі в ЦНС є критичним для розвитку тривожності (Benham etal., 2014). Структура і функція рецепторів ГАМК-А стали предметом ретельного вивчення. Рецептори ГАМК-А є ліганд-керованими хлорид-селективними іонними каналами, кожен із яких являє собою гетероолігомерний білок, що поєднує п’ять субодиниць, які перетинають нейрональну мембрану. Більшість рецепторів ГАМК-А містять дві α-субодиниці, дві β-субодиниці та одну γ-субодиницю (Rudolph etal., 2002). Субодиниці α і β забезпечують зв’язування ГАМК, тоді як γ-субодиниця визначає чутливість до БЗД, оскільки ці препарати зв’язуються в межах ділянки між α- і γ-субодиницями, підвищуючи ймовірність відкриття каналу у відповідь на дію ГАМК (Sigel and Luscher, 2011). Відкриття проникного для хлориду / бікарбонату каналу принаймні двома молекулами ГАМК індукує приплив негативно заряджених іонів хлориду, спричиняючи тимчасове зниження здатності нейронної мембрани проводити потенціали дії, внаслідок чого виникає фазове гальмування нейрона (Olsen, 2018).
Анксіолітичний ефект лікарських засобів, що зв’язуються з рецептором ГАМК-А, пояснюється полегшенням відкриття хлоридних каналів, яке посилює інгібування нейронів у відповідь на ГАМК. Протягом останнього півстоліття було розроблено численні ліганди рецепторів ГАМК-А як терапевтичні засоби, зокрема анксіолітики, міорелаксанти снодійні та протинападові засоби.
Одним із таких анксіолітичних препаратів є етифоксин (6-хлор-2-етиламіно-4-метил-4-феніл-4H-3,1-бензоксазин). Спорідненість етифоксину до хлоридного каналу, пов’язаного з рецептором ГАМК-А, перебуває в мікромолярному діапазоні, тоді як діапазон спорідненості БЗД до цього каналу є наномолярним (Verleye etal., 1999). Етифоксин діє на ГАМК-ергічну передачу, як через прямий вплив на рецептор ГАМК, так і через непрямий, зумовлений синтезом нейростероїдів, що допомагає алостерично модулювати рецептор ГАМК-А (Belelli and Lambert, 2005).
Пряма дія етифоксину на рецептор ГАМК-А
За даними дослідження in vitro, етифоксин може пригнічувати зв’язування специфічного ліганду рецептора ГАМК-А в корі головного мозку гризунів, що свідчить про наявність місць зв’язування етифоксину з рецептором ГАМК-А.. Докази in vivo про функціональні наслідки зв’язування етифоксину з рецептором ГАМК-А (отримані в дослідженнях на тваринних моделях) продемонстрували, що препарат чинить протисудомну дію, опосередковану через рецептор ГАМК-А (Verleye etal., 1999). Інформацію про взаємодію між етифоксином і ГАМК-А рецепторами було отримано завдяки дослідженням зв’язування радіолігандів, які засвідчили, що сайт зв’язування препарату відрізняється від місць зв’язування ГАМК і БЗД (Verleye etal., 2002).
Анксіолітична дія етифоксину, за даними досліджень invivo, не пригнічується флумазенілом, специфічним антагоністом сайту зв’язування БЗД на рецепторі GABA-А, підтверджуючи уявлення про те, що етифоксин і БЗД зв’язуються з різними ділянками. Ця особливість може пояснювати відсутність негативного впливу етифоксину на седативний ефект і пам’ять порівняно з БЗД. Відповідно до даних дослідження рекомбінантних рецепторів ГАМК-А, стимульована етифоксином ГАМК-ергічна передача зберігається за відсутності субодиниць α або γ, отже, саме β-субодиниця рецептора є критичною для зв’язування препарату. У ЦНС наявні кілька підтипів рецептора ГАМК-А, для яких продемонстровано специфічні анатомічні та субклітинні моделі експресії, а також функціонально різні властивості. Очевидна спорідненість рецепторів ГАМК-А до ГАМК та етифоксину залежить від складу субодиниці, точніше, від димерів α-β субодиниці, що утворюють сайти зв’язування агоністів для ГАМК. У цьому контексті етифоксин має вищу спорідненість до рецепторів, що містять субодиниці β2 або β3, ніж субодиниці β1 (Hamon etal., 2003).
Ефекти етифоксину визначаються наявністю субодиниць β, що чітко відрізняє цей анксіолітичний препарат від інших позитивних алостеричних модуляторів рецептора ГАМК-А, як-от БЗД, оскільки активність останніх залежить від природи субодиниць α і γ (Korpi and Sinkkonen, 2006). Зв’язування етифоксину з β-субодиницями сприяє позитивній алостеричній модуляції хлоридних струмів внаслідок відкриття рецепторів ГАМК-А та пов’язано зі збільшенням тривалості й амплітуди струму в разі ненасичених концентрацій ГАМК. Було продемонстровано поведінкові наслідки такої специфічності зв’язування на стресові стимули, зокрема сильніше реагували тварини, у яких виникала надекспресія β2-субодиниці в ЦНС (Verleye etal., 2011). Вони були чутливішими до анксіолітичних і протинападових ефектів етифоксину, що свідчить про позитивну кореляцію між часткою β2-вмісних субодиниць рецептора ГАМК-А та ефективністю препарату (Verleye etal., 2003).
Непряма дія етифоксину на ГАМК-А рецептор
Окрім прямої дії на рецептор ГАМК-А через зв’язування з β-субодиницею, етифоксин може витісняти [3H]-PK11195, високоспецифічний ліганд TSPO, залежно від концентрації (Schlichter etal., 2000). Роль TSPO у процесі синтезу нейростероїдів у нейронах і гліальних клітинах ЦНС, особливо індукованого етифоксином, не з’ясовано. Вважалося, що TSPO розташований у зоні контакту між зовнішньою та внутрішньою мітохондріальними мембранами та є ключовим для транслокації мітохондріального холестерину, який відіграє роль обмежувальної стадії нейростероїдогенезу (Selvaraj and Tu, 2016). PK11195 може стимулювати стероїдогенез навіть без TSPO, що вказує на наявність іншої молекули-мішені (Tuetal., 2015). Етифоксин може збільшувати інтенсивність нейростероїдогенезу через механізм, незалежний від TSPO (DoRego etal., 2015).
Деякі нейростероїди, що виробляються в мітохондріях, взаємодіють із рецептором ГАМК-А. Прегненолон, який синтезується з холестерину в мітохондріях, перетворюється на прогестерон за допомогою 3α-гідроксистероїддегідрогенази. Своєю чергою прогестерон перетворюється в цитоплазмі на його 3α, 5α-похідні завдяки послідовній дії 5α-редуктази та 3α-гідроксистероїддегідрогенази. Найпотужнішими позитивними алостеричними модуляторами рецепторів ГАМК-А є прогестерон, дезоксикортикостерон і метаболіти тестостерону, зокрема алопрегнанолон (Majewska, 1992). Вони збільшують тривалість імпульсу, що відкриває канал ГАМК-А рецептора, а також середній час відкритого стану рецептора (Olsen, 2018). Підвищення рівня алопрегнанолону в мозку після введення етифоксину, яке спостерігалося на тваринних моделях, чинить синергетичний ефект і полегшує ГАМК-ергічну нейротрансмісію, доповнюючи пряму дію етифоксину на β-субодиницю рецептора ГАМК-А (Belelli and Lambert, 2005).
Молекулярний механізм дії етифоксину пояснюється як його прямим впливом на рецептор ГАМК-А, так і непрямим, зумовленим посиленим синтезом нейростероїдів. Такий подвійний механізм дії корелює з анксіолітичним ефектом етифоксину. Оцінювання впливу різних речовин, що блокують центральну ГАМК-ергічну функцію або синтез нейроактивних стероїдів, засвідчило, що зв’язуючись із різними сайтами розпізнавання на рецепторі ГАМК-А, етифоксин і алопрегнанолон виявляють адитивний вплив для посилення інгібіторної ГАМК-ергічної передачі, внаслідок чого відбувається посилення протинападового ефекту (Verleye etal., 2001, 2008).
Етифоксин і серотонінергічна система
У формуванні тривожної поведінки можуть брати участь й інші нейромедіаторні системи, як-от серотонінергічна система. М. Bourin etal. (2010) продемонстрували на тваринній моделі, що анксіолітичний ефект етифоксину модулюється спільним введенням лігандів 5-HT2A. Власне, до анксіолітичного ефекту етифоксину залучений серотонінергічний механізм.
Оскільки етифоксин прямо не взаємодіє із серотонінергічною системою, ці результати свідчать, що анксіолітична активність етифоксину обмежується взаємозв’язком серотонінергічної та ГАМК-ергічної систем.
Вплив етифоксину на симптоми тривожних розладів
вгоруПрямі чи непрямі ГАМК-ергічні механізми, за допомогою яких етифоксин регулює вегетативні реакції, пов’язані з тривожністю, вивчали в низці досліджень. За даними дослідження впливу етифоксину на вегетативні реакції і на стрес в експерименті з тваринною моделлю тривожності (гіпертермія, спричинена стресом, поведінка замерзання, спричинена стресом, і активація моторики товстої кишки), етифоксин послаблював гіпертермію, зменшував прояви поведінки замерзання й частоту скорочень сліпої та ободової кишок у тварин, які зазнали стресових подій. Анксіолітичний ефект етифоксину передбачає послаблення як поведінкових, так і вегетативних ознак тривожності (Verleye and Gillardin, 2004).
На думку дослідників, анксіолітичні властивості етифоксину пов’язані з його впливом на ГАМК-ергічну нейротрансмісію, а не із взаємодією з рецепторами CRF1 і CRF2 (Verleye etal., 2006). Застосування етифоксину сприяє зменшенню гіперзбудливості й тривожності за алкогольної абстиненції (Verleye etal., 2009). Анксіолітична активність як етифоксину, так і габапентину була порівнянною з активністю БЗД діазепаму або серотонінергічного агоніста диметоксийодоамфетаміну гідрохлориду (Bourin and Hascoet, 2012).
Анксіолітична активність етифоксину
вгоруКлінічна фармакологія
Результати дослідження за участю здорових добровольців, метою якого була характеристика фармакокінетичного профілю, продемонстрували, що після введення разової дози етифоксину (150 мг) молекула є надзвичайно біодоступною (приблизно 90 %) і не зв’язується з клітинами крові, проте міцно зв’язується з білками плазми (88–95 %). Етифоксин швидко всмоктується у шлунково-кишковому тракті; час досягнення максимальної концентрації в крові становить 2–3 години. Препарат швидко метаболізується в печінці з утворенням кількох метаболітів, один із яких, діетилетифоксин, є активним. Він може проникати через плацентарний бар’єр. Період напіввиведення етифоксину становить близько 6 годин, а його активного метаболіту — майже 20 годин; виводиться із сечею у вигляді метаболітів, частково — із жовчю (Choi and Kim, 2015).
Клінічна ефективність
Перші дослідження етифоксину в програмі клінічних розробок для отримання дозволу на продаж було проведено за участю пацієнтів із тривожністю та розладами настрою. Етифоксин порівняно із сульпіридом і плацебо ефективно зменшував ознаки тривожності й такі серцево-судинні симптоми, як задишка та тахікардія. Оскільки анксіолітичні ефекти етифоксину на тваринних моделях були найбільш надійно продемонстровані на моделях стресу, клінічні дослідження проводили за участю пацієнтів із розладами адаптації, супроводжуваних підвищеною тривожністю.
У межах багатоцентрового рандомізованого контрольованого подвійного сліпого дослідження, оцінюючи ефективність етифоксину в порівнянні з буспіроном, у 170 пацієнтів із розладами адаптації та тривожністю (≥ 18 балів за шкалою оцінювання тривоги Гамільтона [HAM-A]), учасники отримували або етифоксин (150–200 мг/день; n = 83), або буспірон (15–20 мг/день; n = 87). Первинним результатом був скоригований показник за HAM-A через чотири тижні; вторинними кінцевими точками — показники поліпшення стану та ефективності терапії (зв’язок між анксіолітичною ефективністю та побічними ефектами) за шкалою загального клінічного враження (CGI-I та CGI-E відповідно) (Rickels etal., 1976). Отримані результати підтвердили перевагу етифоксину над буспіроном за середнім показником HAM-A через чотири тижні (p = 0,05). В учасників групи застосування етифоксину був значно кращий показник за CGI-I, ніж за терапії буспіроном, починаючи із 7-го дня лікування (p < 0,001 на 7-й день і p = 0,02 на 14-й і 28-й дні). Показники CGI-Е на 14-й і 28-й дні також були кращими для групи використання етифоксину (p = 0,01 і p = 0,05). Побічні явища (сонливість, запаморочення та головний біль) зафіксовано в 40,7 і 58,6 % пацієнтів, які отримували лікування етифоксином і буспіроном відповідно.
Терапію етифоксином (50 мг тричі на добу; n = 93) і лоразепамом (2 мг/добу, розділені на три приймання: 0,5 мг вранці та вдень і 1,0 мг увечері; n = 96) порівнювали в амбулаторних пацієнтів із розладами адаптації й тривожністю (≥ 20 балів за HAM-A) (Nguyen etal., 2006). Лікування тривало 28 днів. Основним критерієм оцінювання ефективності був скоригований показник за HAM-A на 28-й день. Вторинними кінцевими точками були показники за CGI, шкалою інвалідності Шихана і шкалою для самооцінювання соціальної адаптації. Обидва види лікування були ефективними для зниження показника за HAM-A, починаючи із 7-го дня втручання. Анксіолітичний ефект етифоксину не поступався дії лоразепаму (для показників HAM-A p = 0,0001 на 7-й день і p = 0,0002 на 28-й день). Проте більша кількість осіб відповіла на лікування (загальне зниження показника HAM-A від вихідного рівня до 28-го дня на ≥ 50 %) у групі терапії етифоксином (72 проти 56 %; p = 0,0288).
В обох групах лікування до 28-го дня поліпшився показник CGI, але частота помітного поліпшення (< 3 балів за CGI) була вищою в групі застосування етифоксину (p = 0,022). Показник CGI-Е на 28-й день теж був кращим для етифоксину (p = 0,038). Ефективність у зниженні інвалідизації та поліпшенні соціальної адаптації була порівнянною для двох препаратів. Не виявлено істотної різниці щодо кількості побічних ефектів між групами, які отримували етифоксин або лоразепам. Сонливість фіксували у 10,7 % осіб, які використовували етифоксин, і у 18,7 % — лоразепам. Результати тестів негайного й відстроченого вільного пригадування через 28 днів були порівнянними в обох групах лікування. Симптоми відміни оцінювали через тиждень після припинення лікування. Кількість пацієнтів із тривожністю після припинення лікування була значно більшою (p = 0,034) у групі приймання лоразепаму (вісім осіб), ніж етифоксину (один учасник).
У дослідженні за участю 32 пацієнтів (25 жінок і сім чоловіків віком 20–50 років, середній вік 31 рік) вивчали порівняльну ефективність етифоксину та гідазепаму в комплексній терапії пацієнтів із тривожно-депресивним розладом (Юрьева и др., 2012). Тривалість розладів на момент обстеження варіювала від 1 до 8 років. За комплексної терапії змішаного тривожно-депресивного розладу завдяки характерологічним особливостям астенічного радикалу із додаванням етифоксину і гідазепаму спостерігалася гармонійна редукція симптомів афективних, особистісних і вегетативних розладів, оцінена як суб’єктивно, так і об’єктивно. Клінічно значуще поліпшення було зафіксовано в пацієнтів, які отримували етифоксин (група 1), уже до 7-го дня лікування, а в осіб, які приймали гідазепам (група 2), — до 15-го дня. Повне нівелювання тривожно-депресивної симптоматики та об’єктивних ознак вегетативної дисфункції в пацієнтів групи 1 спостерігалося до 16-го дня лікування, тоді як у групі 2 — до 28-го дня (рис. 1 і 2).
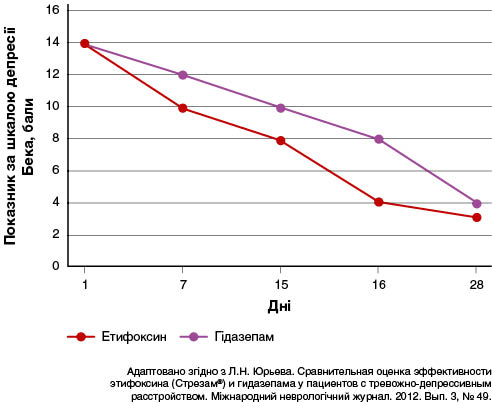
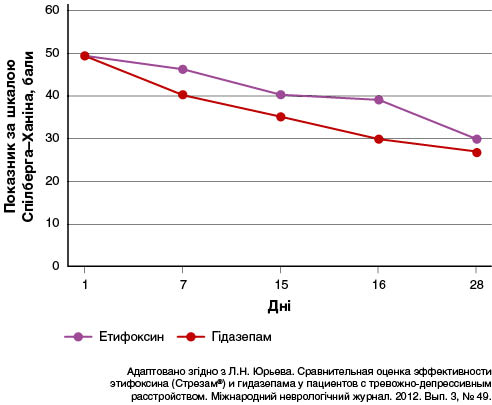
У багатоцентровому подвійному сліпому рандомізованому клінічному дослідженні оцінювали ефективність етифоксину (150 мг/добу; n = 100) порівняно з алпразоламом (1,5 мг/добу; n = 101) у 201 амбулаторного пацієнта з розладами адаптації і тривожністю (≥ 20 балів за HAM-A) (Stein, 2015). Лікування тривало чотири тижні. Первинним результатом був загальний показник за HAM-A на 28-й день, скоригований відповідно до його значення на 1-й день. Вторинними кінцевими точками — показники за шкалою CGI — тяжкість стану (CGI-S) і частота тих, хто відповів на лікування (зниження показника HAM-A на ≥ 50 % між 1 і 28 днями). Терапія як етифоксином, так і алпразоламом сприяла зниженню показників HAM-A на 28-й день, із різницею 1,78 бала між групами лікування на користь застосування алпразоламу (рис. 3).
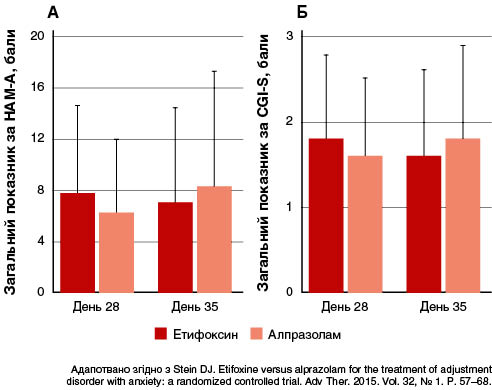
Однак після припинення лікування показники HAM-A продовжували поліпшуватися в групі терапії етифоксину, але підвищувалися в групі приймання алпразоламу. Різниця між цими групами щодо середньої зміни між 28 і 35 днями була достовірною (p = 0,019). На 35-й день вимірювання вторинних результатів продемонструвало подібні результати для етифоксину та алпразоламу.
Більше побічних ефектів, зокрема з боку ЦНС, було пов’язано з лікуванням, у пацієнтів, які отримували алпразолам, а саме значно частіше спостерігалися епізоди сонливості або седації в групі терапії алпразоламом (14 пацієнтів), ніж етифоксином (чотири пацієнти). Підвищену втомлюваність мали лише пацієнти, які отримували алпразолам (чотири особи). Дані цих досліджень демонструють ефективність етифоксину як анксіолітика для пацієнтів із розладами адаптації і тривожністю, хороший профіль переносимості препарату, зокрема щодо збереження когнітивних функцій і пильності. Терапія етифоксином не асоціювалася ні з тривожністю, ні із симптомами відміни після припинення лікування (Stein etal., 2018).
Нейропротекторна дія етифоксину на центральну та периферичну нервову систему
вгоруЗа даними дослідження на тваринних моделях нейрозапалення, ноцицепції та нейродегенерації, етифоксин чинить нейропротекторну дію, чим може частково пояснюватися його сприятливий вплив за неврологічних і психічних розладів (Gazzo etal., 2019). Особливий інтерес становить його активність щодо збільшення синтезу нейротрофічних факторів, залучених до нейропластичності, особливо на синаптичному рівні (Mariga etal., 2017).
Отримані результати додатково свідчать про сприятливий профіль переносимості етифоксину порівняно з БЗД. Зокрема, на моделі ураження периферичних нервів у тварин підтверджено, що етифоксин сприяє регенерації периферичних нервів і росту аксонів, прискорює й поліпшує відновлення рухових функцій, координації та сенсорних функцій (Girard etal., 2008). Етифоксин підтримує синтез та вивільнення гліального нейротрофічного фактора, посилює експресію нейрофіламентів у регенерованих аксонах, поліпшуючи регенерацію сідничного нерва, збільшуючи швидкість нервової провідності та посилюючи експресію нейротрофінів (Zhou etal., 2013). Препарат поліпшує регенерацію сідничного нерва, модулює імунні відповіді та посилює експресію нейротрофіну (Dai etal., 2014). Етифоксин має протизапальні та імуномодулювальні властивості, сприяє зменшенню вироблення прозапальних цитокінів у мікроглії, зокрема інтерлейкінів 1β і 6, фактора некрозу пухлини-α та індуцибельної синтази оксиду азоту. Цей захисний ефект етифоксину значно зменшує неврологічний дефіцит і перигематомний набряк мозку (Lietal., 2017).
У дослідженні на експериментальній моделі розсіяного склерозу було продемонстровано, що етифоксин може зменшити виразність запальної демієлінізації та інфільтрацію периферичних імунних клітин у спинний мозок. Одужання корелювало зі зменшенням запальної патології в поперековому відділі спинного мозку та посиленням олігодендрогліальної регенерації (Daugherty etal., 2013). Імовірно, такий ефект етифоксину може бути опосередкований модуляцією активності TSPO.
На моделі хронічного звуження сідничного нерва етифоксин пригнічував симптоми нейропатичного болю. Цей ефект був повністю опосередкований 3α, 5α-відновленими нейростероїдами та, ймовірно, також алопрегнанолоном, концентрація якого в спинному мозку значно підвищувалася за лікування етифоксином (Aouad etal., 2014).
Висновки
вгоруЕтифоксин є анксіолітичним небензодіазепіновим препаратом із селективністю до β-субодиниці рецептора ГАМК-А. Він має специфічні фармакологічні властивості, зокрема як пряме, так і непряме (через синтез нейростероїдів) полегшення ГАМК-ергічної нейротрансмісії, що сприяє потенційній непрямій серотонінергічної активності. Крім того, етифоксин чинить вплив на процеси запалення імунітету, які, як відомо, пов’язані з тривожністю.
Лікування етифоксином було ефективним для пацієнтів із розладами адаптації й тривожністю в добових дозах 150–200 мг. Профіль переносимості етифоксину кращий, ніж у бензодіазепінів, зокрема через відсутність впливу на пам’ять і пильність, а припинення лікування цим препаратом не призводить до синдрому відміни або відновлення тривожності. Анксіолітична ефективність етифоксину, його хороша переносимість без розвитку лікарської залежності є вагомими аргументами на користь використання вказаного препарату для лікування пацієнтів із розладами адаптації з коморбідною тривожністю.
Підготувала Наталія Купко
Наш журнал
в соцсетях:



Мнения экспертов



Выпуски за 2025 Год
/thumbs/widget_nn25_10-165_titul.jpg)
/thumbs/widget_nn25_9-164_titul.jpg)
/thumbs/widget_nn25_8-163_titul_.jpg)
/thumbs/widget_nn25_7-162_titul-1.jpg)
/thumbs/widget_nn25_6-161_titul.jpg)
/thumbs/widget_nn25_5-160_titul.jpg)
/thumbs/widget_nn25_4-159_titul-1.jpg)
/thumbs/widget_nn25_3-158_titul.jpg)

/thumbs/widget_nn25_2-157_titul-1.jpg)
/thumbs/widget_nn25_1-156_titul-1.jpg)
Содержание выпуска 10 (165), 2025
Содержание выпуска 9 (164), 2025
Содержание выпуска 8 (163), 2025
-
Всесвітній день психічного здоров’я: належна підтримка пацієнтів як суспільний пріоритет
-
Посттравматичний стресовий розлад, спричинений війною: аналіз на основі літературних образів
-
Нейрогенний синдром грудного виходу: пошук оптимального підходу до лікування
-
Минуле та сьогодення Української протиепілептичної ліги: тридцять років складного та успішного шляху
-
Падіння в похилому віці: оцінювання ризику, аспекти профілактики й менеджменту
-
Тернистий шлях застосування бензодіазепінів: від величі до забуття та можливого відродження
-
Досягнення та підтримання стану ремісії в пацієнтів із великим депресивним розладом
Содержание выпуска 7 (162), 2025
-
Минуле та сьогодення Української протиепілептичної ліги: тридцять років складного та успішного шляху
-
Можливості фармакотерапії за уражень мозку, що супроводжуються порушенням свідомості
-
Застосування високих доз оланзапіну в лікуванні пацієнтів із резистентними формами шизофренії
-
Лікування пацієнтів із нейропатичним болем на первинній ланці допомоги
Содержание выпуска 6 (161), 2025
-
Вплив соціальних мереж на психічне здоров’я дітей і підлітків
-
П’ять порад батькам, як знизити вплив соціальних мереж на дитину / підлітка
-
Минуле та сьогодення Української протиепілептичної ліги: тридцять років складного та успішного шляху
-
Діагностична цінність клінічного оцінювання когнітивних функцій
-
Вплив війни на психічне здоров’я молоді: роль резилієнсу та психологічних інтервенцій
-
Фармакотерапія пацієнтів із деменцією: первинна ланка медичної допомоги
-
Методичні рекомендації щодо профілактики професійного вигоряння медичних працівників
Содержание выпуска 5 (160), 2025
-
Поліпшення психологічного стану населення в умовах довготривалої війни
-
Ефективність поетапної програми психологічних втручань для мігрантів
-
Альтернативний підхід до терапії тривожних розладів: важливість правильного титрування дози
-
Аналіз ефективності фармакотерапії депресії у жінок дітородного віку
-
Модель поетапного лікування пацієнтів із ноцицептивним болем
Содержание выпуска 4 (159), 2025
-
Психіатрія способу життя: нові горизонти для психічного здоров’я
-
Поліпшення функціонування як ключова мета лікування пацієнтів із великим депресивним розладом
-
Розлади харчової поведінки: серйозність проблеми та сучасні підходи до її вирішення
-
Антидепресант із мультимодальною дією: можливості застосування міансерину в клінічній практиці
-
Сучасні підходи до діагностування та лікування пацієнтів із кататонією
-
Фармакологічне лікування пацієнтів із шизофренією та пов’язаними з нею психозами
Содержание выпуска 3 (158), 2025
-
Всесвітній день поширення інформації про аутизм: спростовуємо поширені міфи
-
Резистентна до лікування депресія: можливості аугментації терапії
-
Фармакотерапія тривожних розладів і нейропротекція: альтернатива бензодіазепінам
-
Лікування пацієнтів підліткового віку із шизофренією: ефективність і безпека антипсихотичної терапії
-
Лікування депресії в пацієнтів з ішемічною хворобою серця або ризиком її розвитку
-
Профілактична фармакотерапія епізодичного мігренозного головного болю в амбулаторних умовах
-
Стратегії зниження дозування бензодіазепінів: коли ризики переважають користь
Содержание выпуска 1, 2025
-
Когнітивні порушення судинного генезу: діагностування, профілактика та лікування
-
Лікування ажитації за деменції, спричиненої хворобою Альцгеймера
-
Постінсультні нейропсихіатричні ускладнення: типи, патогенез і терапевтичні втручання
-
Перспективи застосування препаратів на основі рослинних компонентів для лікування депресії
-
Застосування диклофенаку за неврологічних станів: перевірена ефективність і пошук нових підходів
-
Постінсультний емоціоналізм: патофізіологія, поширеність та лікування
Содержание выпуска 2 (157), 2025
-
Деякі питання запровадження оцінювання повсякденного функціонування особи
-
Важливість співвідношення «доза-ефект» при застосуванні нестероїдних протизапальних препаратів
-
Медикаментозний паркінсонізм: причини, наслідки та шляхи уникнення
-
Фармакотерапія пацієнтів із шизофренією: важливість поліпшення рівня соціальної залученості
Содержание выпуска 1 (156), 2025
-
Підтримка психічного здоров’я на первинній ланці надання медичної допомоги
-
Лікування депресії в літніх пацієнтів: вплив на патофізіологію розладу, ефективність та безпека
-
Нестероїдні протизапальні препарати: багаторічний досвід та особливості застосування
-
Фармакотерапія великого депресивного розладу: пошук антидепресантів з оптимальною ефективністю
Выпуски текущего года
/thumbs/widget_nn26_5-170_titul.jpg)
/thumbs/widget_nn26_4-169_titul.jpg)
/thumbs/widget_nn26_3-168_titul.jpg)

/thumbs/widget_nn26_2-167_titul.jpg)
/thumbs/widget_nn26_1-166_titul.jpg)
Содержание выпуска 5 (170), 2026
-
Посттравматичний стресовий розлад: розпізнати, зрозуміти, допомогти
-
Диференціальна діагностика та лікування пароксизмальних станів під час війни
-
Лікування больового синдрому в літніх пацієнтів із грижею міжхребцевого диска
-
Диференціальна діагностика міастенії: від флуктуючого птозу до генералізованої слабкості
-
Перинатальна депресія: актуальні алгоритми фармакологічної корекції
-
Алгоритм лікування пацієнтів із шизофренією: міжнародний консенсус INTEGRATE
-
Застосовність фармакотерапевтичних підходів при хронічному безсонні для осіб із гострою інсомнією
Содержание выпуска 4 (169), 2026
-
Індивідуалізована антипсихотична терапія з урахуванням тютюнокуріння
-
Консенсус щодо виявлення й ведення пацієнтів із депресією та коморбідним больовим синдромом
-
Сучасні підходи до застосування кеторолаку при гострому болю в неврологічній практиці
-
Ведення пацієнтів із хворобою Паркінсона: огляд ключових рекомендацій
Содержание выпуска 3 (168), 2026
-
Розширення ролі носимих медичних пристроїв у сучасній неврологічній практиці
-
Роль мистецтва в реабілітації та поліпшенні ментального здоров’я
-
Венлафаксин у терапії ПТСР: від доказової бази до вибору індивідуальної стратегії
-
Нові підходи, сміливі ідеї та найактуальніші теми у сфері неврології
-
Міастенія під маскою депресії та тривоги: клінічні пастки для психіатра і тактика ведення пацієнта
-
Нейропротекція та когнітивна реабілітація в сучасній психіатрії: фокус на комбіновану терапію
-
Антидепресант тразодон: фармакологічний профіль та мультимодальні й мультифункціональні ефекти
-
Рекомендації щодо ведення пацієнтів із нейропатичним болем унаслідок травми спинного мозку
Содержание выпуска 1, 2026
-
Штучний інтелект у геріатричній психіатрії: поєднання точності з людським досвідом
-
Синдром старечої астенії та деменція: клінічні виклики і сучасні рекомендації
-
Реабілітація, відновлення та повернення пацієнтів до активного життя після інсульту
-
Порушення когнітивних функцій, деменція та депресія у літніх осіб
Содержание выпуска 2 (167), 2026
-
Нові підходи, сміливі ідеї та найактуальніші теми у сфері неврології: дебати
-
Нейробореліоз: коли інфекція маскується під неврологічне захворювання
-
Хвороба Вільсона: від неспецифічних скарг до встановлення діагнозу
-
Призначення антипсихотичних препаратів для лікування психічних розладів
-
Підходи до лікування посттравматичного стресового розладу у військових
-
Аспекти реабілітації після інсульту для оптимального функціонального відновлення пацієнтів
Содержание выпуска 1 (166), 2026
-
Психіатрична допомога в Україні у 2025 році: що реально змінилося для лікаря і пацієнта
-
Дулоксетин: лікування депресивних і тривожних розладів та хронічного болю
-
Нові підходи, сміливі ідеї та найактуальніші теми у сфері неврології
-
Нейробіологічні чинники формування терапевтичної дії плацебо та ноцебо у клінічній практиці
Рассылка
Будьте в курсе последних обновлений – подпишитесь на рассылку материалов на Ваш e-mail
Подписаться














